Articles
- Page Path
- HOME > Restor Dent Endod > Volume 49(4); 2024 > Article
-
Research Article
Evaluation of mineral induction ability and cytotoxicity of carbonated hydroxyapatite for pulp tissue regeneration: an
in vitro study -
S. Swathi Priyadharshini1
 , Chinnasamy Ragavendran1, Anand Sherwood2, J. Ramana Ramya3, Jogikalmat Krithikadatta1
, Chinnasamy Ragavendran1, Anand Sherwood2, J. Ramana Ramya3, Jogikalmat Krithikadatta1 -
Restor Dent Endod 2024;49(4):e40.
DOI: https://doi.org/10.5395/rde.2024.49.e40
Published online: October 29, 2024
1Department of Cariology, Saveetha Dental College and Hospitals, Saveetha Institute of Medical and Technical Sciences, Chennai, TN, India.
2Department of Conservative Dentistry and Endodontics, C.S.I. College of Dental Sciences and Research, Madurai, TN, India.
3Department of Periodontics, Saveetha Dental College and Hospitals, Chennai, TN, India.
- Correspondence to Chinnasamy Ragavendran, PhD. Department of Cariology, Saveetha Dental College and Hospitals, 162 Poonamallee High Rd., Velappanchavadi, Chennai, TN 600077, India. ragavan889@gmail.com
• Received: April 2, 2024 • Revised: May 26, 2024 • Accepted: June 11, 2024
Copyright © 2024. The Korean Academy of Conservative Dentistry
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
-
Objectives This study aimed to evaluate carbonated hydroxyapatite (CHA)’s ability for mineral induction and its in vitro cytotoxicity with human dental pulp cells.
-
Materials and Methods Precursors for the study include di-ammonium hydrogen phosphate and calcium nitrate tetrahydrate, with sodium hydrogen carbonate added to achieve different levels of carbonate substitution. The synthesized CHA samples are characterized using X-ray diffraction, Fourier transform infrared spectroscopy, and Raman spectroscopy. Scanning electron microscopy (SEM) was used to observe morphology. For 14 days at 37°C, samples were submerged in simulated body fluid to assess their mineral induction capabilities. SEM was used to confirm apatite formation on sample surfaces. The cytotoxicity assay was used to assess the vitality of the cells following their exposure to various concentrations of CHA.
-
Results The Joint Committee on Powder Diffraction Standards data for HA aligned well with the results from X-ray diffraction analysis of CHA across 3 different concentrations, indicating strong agreement. Fourier transform infrared spectra indicated the presence of phosphate, hydroxyl, and carbonate groups within the samples. SEM and Energy-dispersive X-ray analysis show agglomerated and flaky nanoparticles. All the samples are bioactive, but the formation of apatite differs from one another. In vitro cytotoxicity assay showed that over 70% of cells maintain viability.
-
Conclusions The results of this study may provide insight into the potential use of carbonated HA as a dental pulp-capping material for vital pulp therapy.
INTRODUCTION
The primary objective of pulp treatment is to maintain the integrity and health of the oral tissues [1]. Vital pulp therapy (VPT) is primarily recommended for immature permanent teeth with reversible pulpitis, which can result from mechanical pulp exposure during carious excavation or operative procedures, or from traumatic pulp exposure in healthy teeth with minimal bacterial contamination [2]. Recent findings suggest that VPT can be effectively used not only in deciduous and immature permanent teeth but also in mature permanent teeth with carious pulp exposure, even when clinical signs suggest irreversible pulpitis [3]. VPT in these studies indicates that the procedures used for direct pulp capping or pulpotomy of young permanent teeth with reversible pulpitis might also be effective for mature permanent teeth with irreversible pulpitis. However, it is important to note that treating mature permanent teeth with irreversible pulpitis is not typically considered part of VPT [2].
Direct and indirect pulp capping and pulpotomy are examples of VPT techniques. In these procedures, clinicians place a pulp capping material over affected pulp tissue to maintain its vitality and promote dentin bridge formation. In VPT, the progenitor dental pulp stem cells migrate, differentiate, and form odontoblast-like cells that produce reparative dentin [4]. Ideally, materials used for pulp capping should be inert, non-toxic, and “bio-active” towards tissues, promoting cell migration, proliferation, and differentiation [5].
The ability to regenerate dental pulp is a major area of dental research, as it has the potential to restore the function and integrity of teeth injured by caries or trauma [6]. Prior research has explored the utilization of hydroxyapatite (HA) as a base for tissue regeneration, but there is limited research on the effects of carbonate doping on its biocompatibility and mineral induction properties [7]. Apatite formation signifies the ability of the materials to interact with biological fluids and promote the nucleation and growth of mineral deposits, mimicking the natural process of bone formation [8,9]. By studying a material’s bioactivity in vitro, researchers gain valuable information about its suitability for various medical applications.
In the world of biomedical applications, calcium phosphate shines as a material for bone implants. They effectively replace bone that’s been compromised by disease or injury [10]. The incorporation of carbonate into HA has been found to improve its biological activity by changing its crystal morphology, decreasing crystallinity, increasing solubility, and increasing the local concentration of calcium and phosphate ions [11].
Carbonate ions can substitute within the apatite structure either at the hydroxyl (OH) site (referred to as A-type when synthesized at elevated temperatures) or at the phosphate (PO4) site (referred to as B-type). The closest analogues of biological apatites are type B carbonate [12]. The lack of research on carbonated hydroxyapatite (CHA) and its potential for pulp tissue regeneration represents a significant knowledge gap in the field.
Animal studies have explored the use of CHA as a bone graft material; however, none have assessed its potential for pulp protection [13,14,15,16]. Conducting comparative studies with existing pulp capping materials, such as calcium hydroxide or mineral trioxide aggregate (MTA), can facilitate a better understanding of the relative efficacy and advantages of CHA.
MTA has emerged as a versatile biomaterial with a wide range of applications in endodontics. These applications include direct pulp capping, pulpotomy, apexification, root-end filling, and perforation sealing [17]. MTA is categorized as a hydraulic cement and is composed of tricalcium silicate. MTA exhibits limitations associated with its extended setting time. This necessitates additional clinical procedures and compromises handling characteristics, ultimately leading to delays in achieving the final restoration [18]. Following the introduction of Biodentine (Septodont, Saint-Maur-des-Fosses, France), a calcium silicate cement with the advantage of quicker setting time, challenges persist due to its high cost and longer setting time compared to calcium hydroxide [19]. To address these drawbacks, HA-based materials emerge as a promising alternative, offering excellent biocompatibility [20].
Exploring CHA as an alternative may overcome these drawbacks and offer improved clinical outcomes. The research question is “How does the synthesized and characterized CHA perform in terms of in vitro cell viability when exposed to human dental pulp cells, and what is its capability for mineral induction compared to existing materials?” Hence, the aim of this research is to synthesize and characterize CHA, examining its in vitro cytotoxicity upon exposure to human dental pulp cells, and assessing its capacity for mineral induction.
MATERIALS AND METHODS
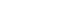
The study protocol was reviewed and authorized by an Institutional Review Board (IRB) with reference number SRB/SDC/PhD/ENDO-2309/23/TH-081. The synthesis of CHA was conducted according to the methodology outlined by Othman et al. [21]. Calcium nitrate tetrahydrate, Ca (NO3)2.4H2O (Merck Life Science Private Limited, Mumbai, Maharashtra, India), is taken as a calcium precursor. A calcium precursor solution was added drop-wise to a solution of the phosphate precursor, diammonium hydrogen phosphate ((NH4)2HPO4) (Merck Life Science Private Limited). CHA synthesis employs sodium hydrogen carbonate (NaHCO3) (Merck Life Science Private Limited) as a source of carbonate ions (CO3
2−). The reaction utilizes calcium and phosphate precursor solutions at a concentration ratio of 1:0.6 M. Ammonium hydroxide, NH4OH (Merck Life Science Private Limited), is used as a pH regulator. Adding 0.05 M, 0.1 M, and 0.5 M of sodium hydrogen carbonate to di-ammonium hydrogen phosphate solution prior to the reaction achieves 3 levels of carbonate substitution. An initial concentration of 1 M was used to prepare all of the solutions.
Firstly, a solution containing phosphate ions was taken in a beaker. The carbonate solution (CO3
2− source) was introduced drop-wise into the phosphate solution with a consistent rate of 40 drops per minute under continuous stirring. To maintain a basic environment (pH ≥ 11), ammonium hydroxide was concomitantly added drop-wise to modulate the pH of the combined solution throughout the process. Following the complete addition of the carbonate solution, the mixture was stirred for an additional 30 minutes. Subsequently, the carbonate-phosphate mixture was added drop-wise to the continuously stirred calcium ion solution. This resulted in a gradual color change of the combined solution, transitioning from transparent to a milky white hue, indicative of precipitate formation.
To achieve a complete reaction, the combined solution was stirred for an additional 30 minutes following thorough mixing. The resulting white precipitate was subsequently washed 3 times with deionized water to eliminate residual impurities. The precipitate was then oven-dried at 100°C for 24 hours. Finally, the dried material was pulverized using an agate mortar and pestle before sieving through a 90 μm mesh (Figure 1). The aforementioned methodology was employed to prepare 3 distinct carbonate solution concentrations for subsequent characterization.
The crystalline phases of the samples were identified using X-ray diffraction (XRD) (D8 ADVANCE, Bruker-AXS, Karlsruhe, Germany). Fourier-transform infrared spectroscopy (FTIR) (ALPHA, Bruker, Billerica, MA, USA) was then employed to characterize the molecular structure of the samples. Raman spectroscopy (BRUKER RFS27: Standalone FT) was employed to determine the crystallographic orientation of a sample. Scanning electron microscopy (SEM) and energy-dispersive X-ray analysis (EDAX) (JEOL IT-800SHL, JEOL, Tokyo, Japan) were used to observe the morphology and the elemental composition of the obtained CHA’s.
An in vitro bioactivity test was conducted using simulated body fluid (SBF) following the established protocol by Kokubo et al. [22]. The samples under investigation were prepared in pelleted form, likely to ensure uniformity and ease of handling during testing. The prepared samples were subsequently immersed in 10 mL of SBF solution within a flat-bottomed container. The use of a flat-bottomed container ensures maximum surface contact between the samples and the SBF solution, facilitating effective interaction. The samples underwent immersion in 10 mL of SBF solution for a period of 14 days maintained at 37°C in an incubator. This temperature closely resembles that of the human body, ensuring that the test conditions closely mimic physiological conditions. To simulate dynamic physiological conditions, the SBF solution was replenished every other day throughout the 14-day immersion period. This periodic replacement helps to maintain the chemical balance of the solution, ensuring that it remains conducive to apatite formation throughout the test. At the end of the 14-day immersion period, the samples were retrieved from the SBF solution and subsequently dried. To confirm the formation of apatite on the surface of the samples, SEM analysis was subsequently performed.
Human dental pulp cells were separated from normal adult teeth, aged between 18 and 25 years old that were extracted for orthodontic purposes at Saveetha Dental College and Hospital. The isolation of human dental pulp cells was carried out through enzymatic digestion using collagenase (900 U/mL) and dispase (400 U/mL) at 37°C for 1 hour. Primary human dental pulp cell cultures were established using Rosewell Park Memorial Institute (RPMI) 1640 medium (Invitrogen Corporation, Carlsbad, CA, USA) supplemented with 20% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cultures were kept at 37°C in a humidified atmosphere with 5% CO2. Cells were sub-cultured at around 80% confluence, and the culture medium was changed every 3 days. For every subsequent in vitro experiment, cells of passage 2 were used.
In accordance with ISO 10993-12, material preparation involved incubating 500 mg samples (control and experimental) with 1 mL of RPMI medium at a ratio of 0.2 g/mL. This mimicked an extraction ratio of 0.2 g material per 1 mL medium. The incubation occurred at 37°C in a humidified incubator with 5% CO2 for 24 hours. Following incubation, the culture medium containing the material extracts was sterilized by filtration through 0.22 µm cellulose acetate filters (MilliporeSigma, St. Louis, MO, USA) for subsequent cytotoxicity assessment using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay.
This study aimed to assess the cytotoxicity of CHA at varying concentrations (0.05, 0.1 and 0.5 M) on human dental pulp cells using the MTT assay, following the protocol established by Koka et al. [23]. Briefly, cells from human dental pulp were seeded at a density of 1 × 104 cells/well onto 96-well plates, allowed to adhere, and then exposed to various concentrations of CHA. After a 24-hour incubation, the culture medium was replaced with 10 μL of MTT solution (10 mg/mL), followed by a further 4-hour incubation with the cells. After dissolving the formazan crystals in 100 μL of dimethyl sulfoxide, the absorbance of the crystals was measured at 570 nm using a Synergy Hybrid Multi-Mode Reader. The following formula was then used to determine cell viability:
The experiments were performed in triplicate to ensure data reproducibility. Data are presented as the mean ± standard error of the mean, representing independent observations. Statistical analysis was carried out using SPSS version 13 software (SPSS, Chicago, IL, USA). One-way analysis of variance with Tukey's post hoc multiple comparison test was employed to assess for statistically significant differences between groups. A p value of less than 0.05 was considered statistically significant.
RESULTS
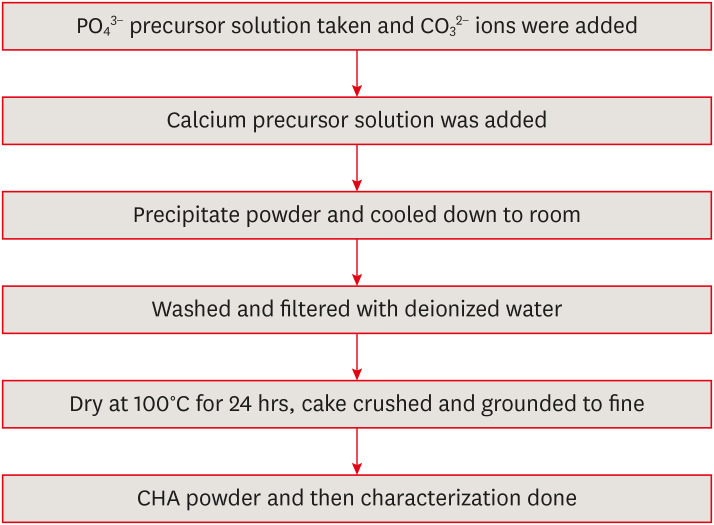
The XRD patterns of the synthesized CHA at the 3 investigated concentrations are presented in Figure 2. The XRD patterns of CHA with concentrations of 0.05, 0.1 and 0.5 M were similar to the HA (09-0432) The Joint Committee on Powder Diffraction Standards (JCPDS) data. Peaks at 25.8°, 28.1°, 29.0°, 31.7°, 32.2°, 32.8°, 34.0°, 39.7°, 46.6°, 48.1°, 49.4°, 53.1°, and 55.9° matched the crystal lattice reflections of HA: (002), (102), (210), (211), (112), (300), (202), (310), (222), (312), (213), (004), and (322).
Figure 2
X-ray diffraction patterns of (a) 0.05 M carbonated hydroxyapatite (CHA), (b) 0.1 M CHA, and (c) 0.5 M CHA.
Lattice parameters and crystallite size of the samples were determined using MAUD software (Trento, Italy). The results are presented in Table 1. On increasing the carbonate concentration in the HA matrix, the lattice constant ‘a’ value reduced with an increase along the c-axis. The lattice parameter c/a ratio was calculated as 0.72, 0.98, and 1.09, respectively, for 0.05 CHA, 0.1 CHA, and 0.5 CHA.
Table 1
Lattice parameters and crystallite size of (a) 0.05 CHAp, (b) 0.1 CHAp, and (c) 0.5 CHAp
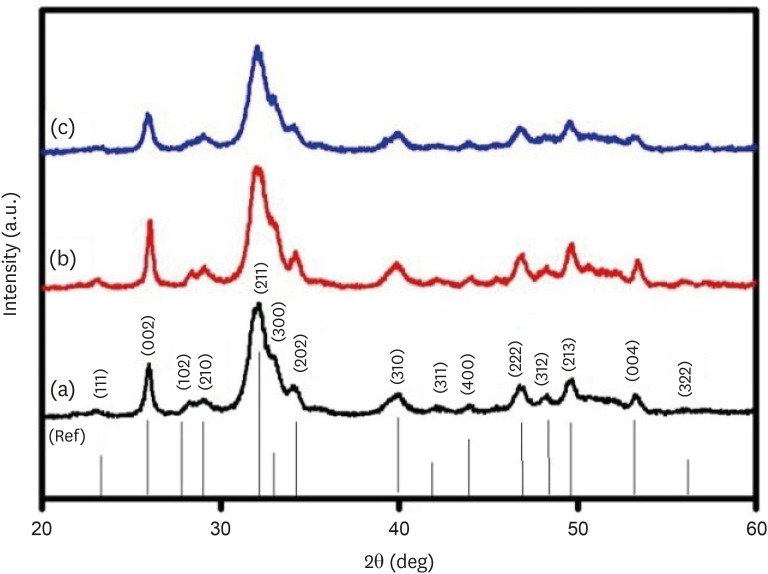
The FTIR spectra of the CHA samples at various concentrations are shown in Figure 3. The FTIR spectra demonstrate the presence of groups such as phosphate (PO4
3−), hydroxyl (OH−), and carbonate (CO3
2−) in the samples. Figure 3B and 3C provide an enlarged view of the regions from 1,300–1,600 cm−1 and 1,000–800 cm−1, respectively. The low-intensity peaks observed in the range of 1,350–1,550 cm−1 correspond to the ν3 asymmetric stretching modes of carbonate groups. The peaks at 1,416, 1,462, and 873 cm−1 show B-type carbonated apatite presence. It means CO3
2− groups replace PO4
3− groups in the HAP lattice. The bands at 1,022 and 1,090 cm−1 arise from the ν3 triply degenerate asymmetric stretching vibration of phosphate. The ν1 non-degenerate symmetric stretching vibration leads to the phosphate bands observed at 962 cm−1. The peaks at 603 and 567 cm−1 are attributed to the ν4 triply degenerate bending vibration of the PO4
3− group. The distinct and slender band observed at 630–670 cm−1 confirms the presence of the apatite phase and is linked to the hydroxyl group. As the concentration of carbonate increased, the intensity of peaks at 1,462 and 1,416 cm−1, attributed to ν3a and ν3b vibrations, respectively, increased. At high concentrations, new peaks at 1,340 and 1,550 cm−1 were also observed.
Figure 3
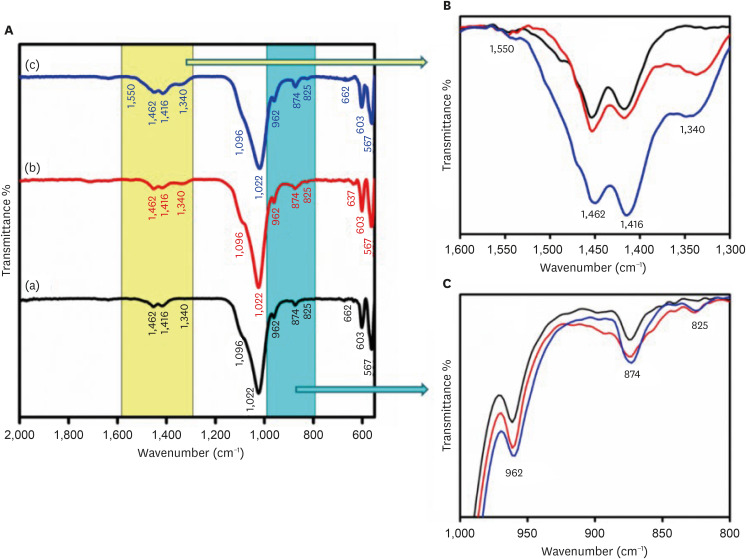
(A) Fourier-transform infrared spectroscopy spectra of (a) 0.05 M carbonated hydroxyapatite (CHA), (b) 0.1 M CHA, and (c) 0.5 M CHA. (B) Magnification of the region 1,600 cm−1–1,300 cm−1. (C) Magnification of the region 1,000 cm−1–800 cm−1.
The Raman analysis of the samples was carried out, and the Raman spectra were plotted for the samples as shown in Figure 4. Raman spectroscopy confirmed the presence of the characteristic vibrational modes associated with the PO4
3− group in the samples. These include the ν1, ν2, ν3, and ν4 bands. Notably, the peak observed at 430 cm−1 corresponds to the ν2 symmetric stretching vibration of the phosphate group, arising from doubly degenerate O-P-O bending modes with E symmetry. The band at 583 cm−1 corresponds to the ν4 bending modes of the PO4
3− group owing to triply degenerated T2 modes of O-P-O bending. The band at 959 cm−1 is associated with the ν1symmetric stretching motion (A1 symmetry) of the P-O group, while the peak within the range of 1,008–1,055 cm−1 corresponds to the triply degenerated T2 mode (asymmetric P-O stretching) of the O-P-O unit (ν3) present in the PO4
3− group. The peak at 1,067 cm−1 is characteristic of type B carbonated apatite and is responsible for the ν1 symmetric stretching of the CO3
2− group.
Figure 4
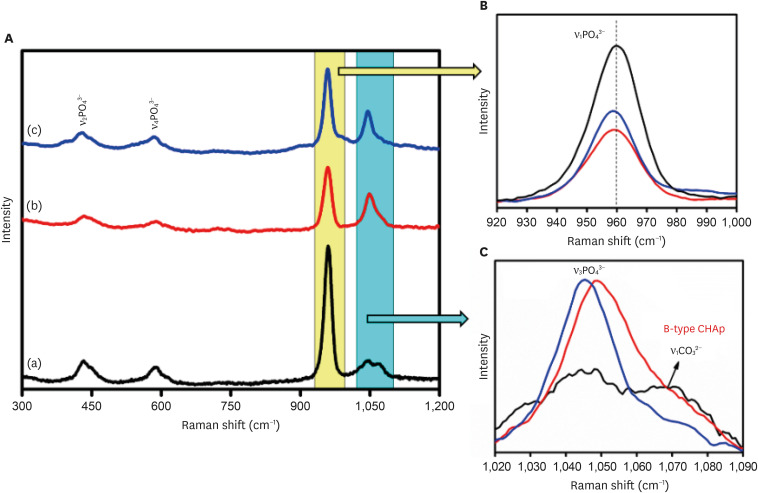
(A) Raman spectra of (a) 0.05 M carbonated hydroxyapatite (CHA), (b) 0.1 M CHA, and (c) 0.5 M CHA. (B) Magnification of the region 920 cm−1–1,000 cm−1. (C) Magnification of the region 1,020 cm−1–1,090 cm−1.
CHAp, carbonated hydroxyapatite powder.
The SEM micrographs and EDAX spectra of the CHA samples are shown in Figure 5A, 5C, 5E and Figure 5B, 5D, 5F, respectively. The CHA prepared by the wet-chemical method exhibited agglomerated and flaky nanoparticles. The nanopores were observed on the surface of the agglomerated flakes (Figure 5C). The EDAX spectra revealed the presence of calcium, phosphate, and carbon. The concentration of carbon increased and the concentration of calcium ions decreased with increasing carbonate concentrations.
Figure 5
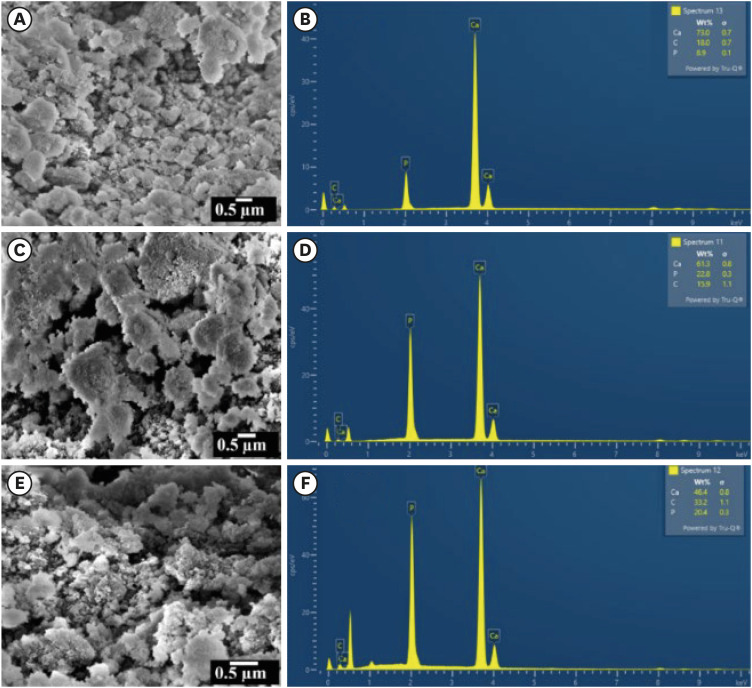
Scanning electron microscopy micrographs and energy-dispersive X-ray analysis spectra for specimens labelled as (A, B) 0.05 M carbonated hydroxyapatite (CHA), (C, D) 0.1 M CHA, and (E, F) 0.5 M CHA.
The SEM micrographs of the samples after being immersed in the SBF for 14 days showed all the samples are bioactive but the formation of apatite differs from one another (Figure 6). The HA showed the apatite layer deposition on the pellet surface and the EDAX confirmed the presence of Ca and P (Figure 6A). The 0.05CHA sample showed less deposition of apatite on the surface (Figure 6B). On increasing the concentration of carbonate to 0.1 M CHA, the apatite deposition is enhanced (Figure 6C). Figure 6E and 6F exhibit the formation of uniform spherical apatite on the surface of commercially available MTA and a uniform rod-like structure on the surface of Biodentine. EDAX analysis conclusively identified the presence of calcium (Ca), phosphorus (P), and oxygen (O) in the precipitates formed on all specimens.
Figure 6
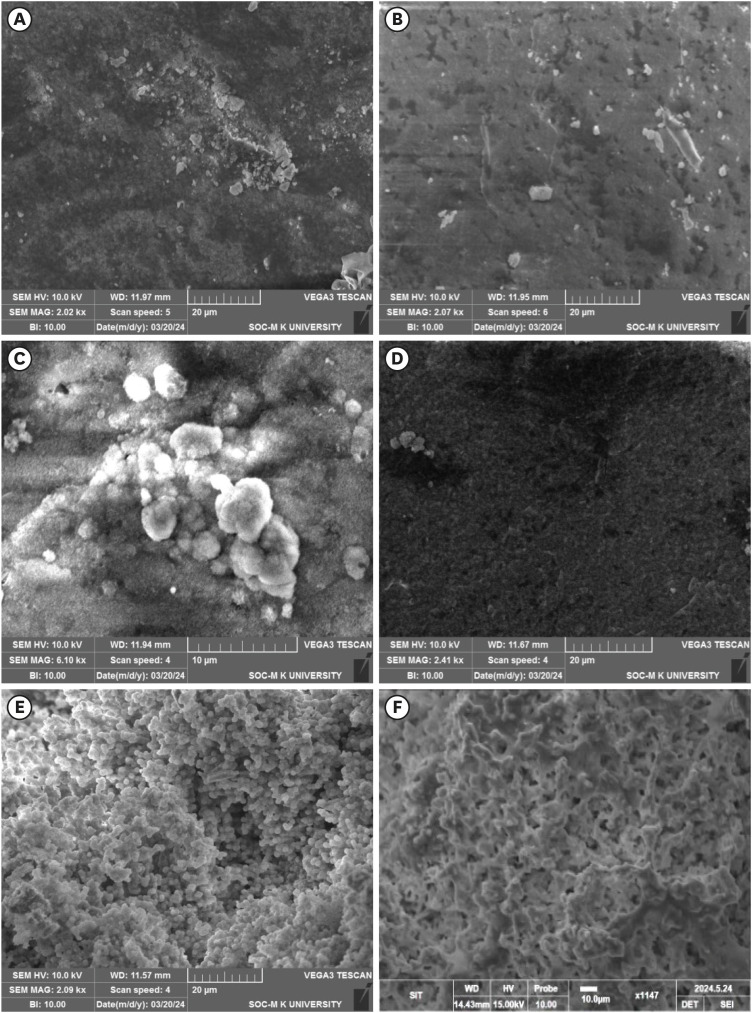
Scanning electron microscopy micrographs of the simulated body fluid-soaked sample with apatite deposited on the surface of (A) hydroxyapatite, (B) 0.05 carbonated hydroxyapatite (CHA), (C) 0.1 CHA, (D) 0.5 CHA, (E) mineral trioxide aggregate, and (F) Biodentine.
In testing cytotoxicity in vitro, colorimetric assays for mitochondrial activity (MTT) indicated that over 70% of cells maintained viability in all 3 concentrations tested. No incorporation of CHA powder samples was observed in human dental pulp cells cultured for 24 hours. Figure 7 shows in vitro cytotoxicity of human Dental pulp stem cell lines after culturing in the synthesized CHA samples along with positive and negative control determined over 24 hours by MTT assay.
Figure 7
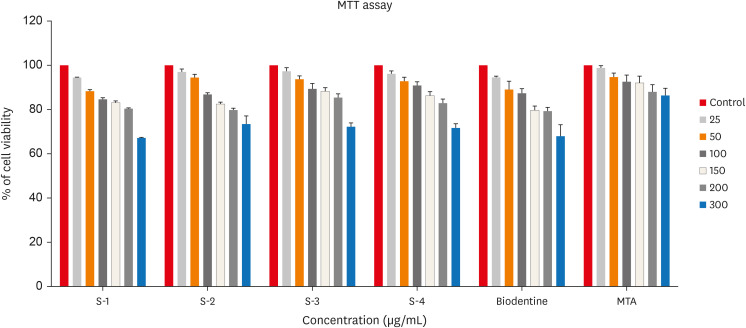
In vitro cytotoxicity of human Dental pulp stem cell lines after culturing in the synthesized carbonated hydroxyapatite (CHA) samples along with positive and negative control determined over 24 hours by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. 0.05 M CHA (S-1), 0.5 M CHA (S-2), 0.1 M CHA (S-3), hydroxyapatite (S-4), Biodentine, mineral trioxide aggregate (MTA).
DISCUSSION
This investigation aims to assess the in vitro cytotoxicity and mineral induction potential of CHA as a material for dental pulp tissue regeneration. The results of this study showed that CHA material exhibited good cell viability and mineral induction properties, making it a suitable material for pulp tissue regeneration. Previous studies have shown that HA has been widely used in dental and orthopedic applications due to its biocompatibility and ability to stimulate bone growth [24,25]. This in vitro study contributes to the development of new biomaterials for pulp tissue regeneration, which can potentially improve treatment outcomes for those undergoing vital pulp therapies. The in vitro cytotoxicity and mineral induction capability of CHA material were compared to existing materials used for pulp tissue regeneration, such as MTA and Biodentine.
This study employed a precipitation method to synthesize B-type carbonated CHA’s with varying carbonate content. Othman et al. [21] reported that reliable synthesis of a single-phase CHA was only achievable using calcium nitrate tetrahydrate as a precursor, while calcium hydroxide and calcium carbonate were found to be less suitable for this purpose. Therefore, for this study, calcium nitrate tetrahydrate was chosen as the calcium precursor. The precipitation method was chosen for its established advantages, including rapid synthesis, operational simplicity, and the ability to precisely control both the composition and particle size of the resulting material [26]. Hence, the same method was employed in this study for synthesizing the B-type CHA.
In B-type carbonated apatites, changes in lattice parameters involve a reduction in the ‘a’ lattice constant and an increase along the c-axis due to the larger size of PO4 tetrahedra compared to CO3 triangles. This substitution impacts CHA crystallinity, reduces it, and enhances the solubility of CO3 ions within the apatite lattice [26]. The findings in Table 1 illustrate a decrease in the ‘a’ lattice constant and a slight increase along the c-axis, indicating the substitution of smaller carbonate ions for larger phosphate groups. Increasing carbonate concentration in the HA matrix led to a reduction in the ‘a’ lattice constant and an increase along the c-axis [27]. Consequently, the average crystallite size decreased by 33% in 0.5 M CHA compared to lower carbonate concentrations.
As shown in Figure 2, the XRD patterns of the 0.05 M CHA, 0.1 M CHA, and 0.5 M CHA concentrations align well with the JCPDS data for HA (09-0432), indicating agreement with both HA and CHA characteristic peaks within the 31°C–34°C range [26]. This study revealed the absence of secondary phases, confirming the successful synthesis of CHA. The broad peaks observed in the XRD pattern are indicative of the low crystallinity and likely nanocrystalline nature of the precipitated CHA.
The FTIR spectrum displayed in Figure 3 exhibits the characteristic absorption bands of HA. It’s known that the ν3 asymmetric stretching vibrations of carbonate groups split into ν3a and ν3b peaks. The specific positions of these peaks allow for the identification of the carbonate substitution sites within the HA lattice, whether replacing (OH−) or (PO4
3−) [26]. In this study, all 3 samples exhibit peaks at 1,416, 1,453, and 873 cm−1, affirming the synthesis of B-type CHA where carbonate replaces phosphate ions within the HA lattice. As carbonate concentration increases, there’s a tendency for the carbonate apatite type to transition towards the AB type. At higher concentrations (from 0.1 M to 0.5 M), the intensity of peaks at 1,462 (ν3a) and 1,416 (ν3b) increases. At the highest concentration, additional peaks at 1,340 and 1,550 cm−1 emerge, possibly due to excess carbonate ion substitution on the phosphate group. Furthermore, a characteristic peak at 874 cm−1 representing the ν2 out-of-plane bending vibration of carbonate groups shows increased intensity in the 0.5 M CHA sample.
This study evaluated all 3 samples using Raman spectra. At a lower concentration of carbonate ions (0.05 M) in the HA matrix, the intensity of ν1 vibrations is high (Figure 4B). However, as the carbonate concentration increases to 0.1 M, the intensity reduces due to the replacement of CO3
2− ions over PO4
3− ions. Upon further increasing the carbonate ions to 0.5 M, the intensity slightly exceeds that of the 0.1 M CHA, suggesting no further encouragement for occupancy of CO3
2− groups beyond this threshold limit. The ν3 asymmetric stretching modes of the PO4
3− group at 1,048 cm−1 and the ν1 symmetric stretching of the CO3
2− group at 1,070 cm−1 are clearly observed in the 0.05 M CHA (Figure 4C). However, in the 0.1 M CHA, the ν3 vibration of PO4
3− overlaps with a B-type carbonate peak emerging around 1,070 cm−1, causing peak broadening. Additionally, in the 0.5 M CHA, there’s a red-shift towards lower angles in the peak of ν3 asymmetric stretching modes of the PO4
3− group (from 1,048 cm−1 to 1,045 cm−1) due to reduced particle size, and the ν1 symmetric stretching of the CO3
2− group vanished. Figure 5 presents SEM micrographs of the synthesized CHA prepared via precipitation. The micrograph depicts highly agglomerated spherical particles of nano-meter size. The observed carbonate substitution sites are consistent with the findings reported by Benataya et al. [28].
When a material like HA is submerged in SBF, a biomimetic mineralization process ensues. SBF, emulating the ionic composition of human blood plasma, provides an environment conducive to apatite formation [29]. Initially, ions from SBF, including calcium, phosphate, and carbonate, adhere to the material’s surface, creating nucleation sites. These sites facilitate the aggregation of ions, leading to the formation of small apatite clusters or nuclei [30,31]. Prolonged immersion in SBF facilitates the progressive deposition of ions onto the surface. This sustained process promotes the nucleation and subsequent growth of these ions, ultimately leading to the formation of a continuous apatite layer [32].
The 0.05 M CHA sample exhibited reduced apatite deposition on its surface (Figure 6B). Lower concentrations of CHA can result in slower apatite precipitation rates. Consequently, within the same immersion period, less apatite may accumulate compared to higher concentrations where precipitation occurs more rapidly. Upon increasing the concentration to 0.1 M CHA, apatite deposition is enhanced (Figure 6C). Elevating the carbon content to 0.5 M can result in carbon incorporation into the calcium sites of the HA lattice and material carbonization (Figure 6D). This carbon integration disrupts the crystal structure of HA, as carbon atoms possess different sizes and properties compared to calcium ions [33,34]. Therefore, the increased carbon content interferes with the normal mineralization process, leading to a less pronounced apatite layer formation. The surface of commercially available MTA shows the production of homogeneous spherical apatite whereas Biodentine exhibits a uniform rod-like structure on its surface, as illustrated in Figure 6E and 6F. The precipitation occurs preferentially on the surface of the MTA and Biodentine due to the high local concentration of calcium ions. Morita et al. [17] reported a lower mineral induction ability for resin-modified MTA cement compared to conventional MTA cements.
In this study, human dental pulp cells were chosen for in vitro analyses due to their relevance in interacting with biomaterials during reparative dentine formation. Figure 7 demonstrates that CHA exhibits acceptable cell viability, promoting better cell growth compared to both the control group and HA sample, as observed in the morphological behavior reported in vivo and in vitro [35]. Notably, Dos Anjos et al. [36] reported minimal cytotoxic effects on murine pre-osteoblast cells even at high carbonate concentrations within HA.
This study’s significance lies in evaluating cytotoxicity using human dental pulp cells, distinct from the majority of reported studies that focused on osteoblasts/pre-osteoblasts. Previous findings have established CHA as biocompatible with osteoblastic cells [13,25]. Here, 3 different concentrations of CHA (0.05 M, 0.1 M, and 0.5 M) exhibited acceptable cell viability on human dental pulp cells.
CHA exhibits several properties, such as biocompatibility, osteo-inductiveness, osteo-conductiveness, and an accelerated biodegradation rate [25]. In research by Landi et al. [14], porous CHA showed more resorption and more new bone start than porous HA in a distal metaphyseal model of the rabbit based on qualitative X-ray examination. We conducted the synthesis and characterization of B-type CHA at 3 different concentrations. Subsequently, we assessed its cytotoxicity using MTT assays on human dental pulp stem cells and examined its capability to induce apatite formation on the surface. This pre-clinical investigation aimed to explore the potential of CHA in endodontic applications.
A primary limitation of the present study is its preclinical nature. Future in vivo investigations using animal models are warranted to assess the efficacy of this material as a pulp capping agent. Limitation of synthesizing CHA using the precipitation method often yields products with inadequate uniformity, prone to agglomeration can be influenced by various factors such as the concentration of the reactants, the reaction time, and the stirring speed [37]. In terms of its cytotoxicity with pulp cells, it is important to note that the MTT assay is just 1 method of evaluating cell viability and should be used in conjunction with other assays to obtain a more complete understanding of cell behavior. It is important to acknowledge that the MTT assay results can be susceptible to variations arising from inherent factors within the CHA material itself, such as its composition, as well as the specific cell culture conditions employed during the assay [38].
Both MTA and Biodentine have their drawbacks. MTA has prolonged setting times and difficult handling, while Biodentine, although effective, is costlier and takes longer to set than calcium hydroxide [17,19]. Compared to materials like MTA and Biodentine, CHA offers a more biomimetic environment for tissue repair and regeneration because it closely matches the mineral phase of actual bone tissue [14]. This research aims to determine the practical feasibility of using CHA as a pulp capping material in VPT. While laboratory studies have shown promising results, animal tests are still ongoing to evaluate its biocompatibility for pulp capping. Ultimately, clinical trials will be necessary to definitively define CHA’s role as a pulp capping agent.
CONCLUSIONS
The results of this in vitro study demonstrated that CHA has the potential for pulp tissue regeneration due to its mineral induction ability and acceptable cell viability. Future studies could focus on investigating the efficacy of CHA in vivo and exploring its potential applications in pulp tissue regeneration.
-
Conflict of Interest: No potential conflict of interest relevant to this article was reported.
-
Author Contributions:
Conceptualization: Priyadharshini SS, Ragavendran C, Ramya JR.
Data curation: Priyadharshini SS, Ragavendran C.
Investigation: Ramya JR, Sherwood A.
Methodology: Krithikadatta J.
Project administration: Krithikadatta J.
Validation: Ragavendran C, Sherwood A, Krithikadatta J.
Writing - original draft: Priyadharshini SS.
Writing - review & editing: Priyadharshini SS.
- 1. Fuks AB. Current concepts in vital primary pulp therapy. Eur J Paediatr Dent 2002;3:115-120.PubMed
- 2. Lin LM, Ricucci D, Saoud TM, Sigurdsson A, Kahler B. Vital pulp therapy of mature permanent teeth with irreversible pulpitis from the perspective of pulp biology. Aust Endod J 2020;46:154-166.ArticlePubMedPDF
- 3. El-Ma’aita AM, Qualtrough AJ, Darcey J, Hunter MJ. Vital pulp therapy: the past, present and future. Dent Update 2022;49:905-910.Article
- 4. Saghiri MA, Asatourian A, Garcia-Godoy F, Sheibani N. Effect of biomaterials on angiogenesis during vital pulp therapy. Dent Mater J 2016;35:701-709.ArticlePubMedPMC
- 5. Pedano MS, Li X, Yoshihara K, Landuyt KV, Van Meerbeek B. Cytotoxicity and bioactivity of dental pulp-capping agents towards human tooth-pulp cells: a systematic review of in-vitro studies and meta-analysis of randomized and controlled clinical trials. Materials (Basel) 2020;13:2670.PubMedPMC
- 6. Zhang W, Yelick PC. Vital pulp therapy-current progress of dental pulp regeneration and revascularization. Int J Dent 2010;2010:856087.ArticlePubMedPMCPDF
- 7. Radulescu DE, Vasile OR, Andronescu E, Ficai A. Latest research of doped hydroxyapatite for bone tissue engineering. Int J Mol Sci 2023;24:13157.ArticlePubMedPMC
- 8. Miyaza T, Kim HM, Kokubo T, Ohtsuki C, Kato H, Nakamura T. Mechanism of bonelike apatite formation on bioactive tantalum metal in a simulated body fluid. Biomaterials 2002;23:827-832.ArticlePubMed
- 9. Pan H, Zhao X, Darvell BW, Lu WW. Apatite-formation ability--predictor of “bioactivity”? Acta Biomater 2010;6:4181-4188.ArticlePubMed
- 10. Muhammad Syazwan MN, Ahmad-Fauzi MN, Yanny Marliana BI. Co-Sr doped carbonated hydroxyapatite: a biomaterial with enhanced mechanical and bioactivity properties. J Phys Conf Ser 2018;1082:012076.ArticlePDF
- 11. Bang LT, Long BD, Othman R. Carbonate hydroxyapatite and silicon-substituted carbonate hydroxyapatite: synthesis, mechanical properties, and solubility evaluations. Sci World J 2014;2014:969876.ArticlePDF
- 12. Leventouri T, Chakoumakos BC, Papanearchou N, Perdikatsis V. Comparison of crystal structure parameters of natural and synthetic apatites from neutron powder diffraction. J Mater Res 2001;16:2600-2606.Article
- 13. Calasans-Maia MD, Melo BR, Alves AT, Resende RF, Louro RS, Sartoretto SC, et al. Cytocompatibility and biocompatibility of nanostructured carbonated hydroxyapatite spheres for bone repair. J Appl Oral Sci 2015;23:599-608.ArticlePubMedPMC
- 14. Landi E, Celotti G, Logroscino G, Tampieri A. Carbonated hydroxyapatite as bone substitute. J Eur Ceram Soc 2003;23:2931-2937.Article
- 15. Carmo AB, Sartoretto SC, Alves AT, Granjeiro JM, Miguel FB, Calasans-Maia J, et al. Alveolar bone repair with strontium- containing nanostructured carbonated hydroxyapatite. J Appl Oral Sci 2018;26:e20170084.ArticlePubMedPMC
- 16. Habibovic P, Kruyt MC, Juhl MV, Clyens S, Martinetti R, Dolcini L, et al. Comparative in vivo study of six hydroxyapatite-based bone graft substitutes. J Orthop Res 2008;26:1363-1370.PubMed
- 17. Morita M, Kitagawa H, Nakayama K, Kitagawa R, Yamaguchi S, Imazato S. Antibacterial activities and mineral induction abilities of proprietary MTA cements. Dent Mater J 2021;40:297-303.ArticlePubMed
- 18. Voicu G, Didilescu AC, Stoian AB, Dumitriu C, Greabu M, Andrei M. Mineralogical and microstructural characteristics of two dental pulp capping materials. Materials (Basel) 2019;12:1772.ArticlePubMedPMC
- 19. Barczak K, Palczewska-Komsa M, Sikora M, Buczkowska-Radlińska J. Biodentine™–use in dentistry. Literature review. Pomeranian J Life Sci 2020;66:39-45.ArticlePDF
- 20. Fiume E, Magnaterra G, Rahdar A, Verné E, Baino F. Hydroxyapatite for biomedical applications: a short overview. Ceramics 2021;4:542-563.Article
- 21. Othman R, Mustafa Z, Loon CW, Noor AF. Effect of calcium precursors and pH on the precipitation of carbonated hydroxyapatite. Procedia Chem 2016;19:539-545.Article
- 22. Kokubo T, Takadama H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006;27:2907-2915.ArticlePubMed
- 23. Koka P, Mundre RS, Rangarajan R, Chandramohan Y, Subramanian RK, Dhanasekaran A. Uncoupling Warburg effect and stemness in CD133+ve cancer stem cells from Saos-2 (osteosarcoma) cell line under hypoxia. Mol Biol Rep 2018;45:1653-1662.ArticlePubMedPDF
- 24. Szcześ A, Hołysz L, Chibowski E. Synthesis of hydroxyapatite for biomedical applications. Adv Colloid Interface Sci 2017;249:321-330.ArticlePubMed
- 25. Rupani A, Hidalgo-Bastida LA, Rutten F, Dent A, Turner I, Cartmell S. Osteoblast activity on carbonated hydroxyapatite. J Biomed Mater Res A 2012;100A:1089-1096.Article
- 26. Siddiqi SA, Azhar U. Carbonate substituted hydroxyapatite. In: Khan AS, Chaudhry AA, editors. Handbook of ionic substituted hydroxyapatites. Amsterdam: Elsevier; 2019. p. 149-173.
- 27. Madupalli H, Pavan B, Tecklenburg MM. Carbonate substitution in the mineral component of bone: discriminating the structural changes, simultaneously imposed by carbonate in A and B sites of apatite. J Solid State Chem 2017;255:27-35.ArticlePubMedPMC
- 28. Benataya K, Lakrat M, Elansari LL, Mejdoubi E. Synthesis of B-type carbonated hydroxyapatite by a new dissolution-precipitation method. Mater Today Proc 2020;31:S83-S88.Article
- 29. Miyajima H, Touji H, Iijima K. Hydroxyapatite particles from simulated body fluids with different pH and their effects on mesenchymal stem cells. Nanomaterials (Basel) 2021;11:2517.ArticlePubMedPMC
- 30. Edén M. Structure and formation of amorphous calcium phosphate and its role as surface layer of nanocrystalline apatite: Implications for bone mineralization. Materialia 2021;17:101107.Article
- 31. Hong MH, Lee JH, Jung HS, Shin H, Shin H. Biomineralization of bone tissue: calcium phosphate-based inorganics in collagen fibrillar organic matrices. Biomater Res 2022;26:42.ArticlePubMedPMCPDF
- 32. Shin K, Acri T, Geary S, Salem AK. Biomimetic mineralization of biomaterials using simulated body fluids for bone tissue engineering and regenerative medicine. Tissue Eng Part A 2017;23:1169-1180.PubMedPMC
- 33. Yang WH, Xi XF, Li JF, Cai KY. Comparison of crystal structure between carbonated hydroxyapatite and natural bone apatite with theoretical calculation. Asian J Chem 2013;25:3673-3678.Article
- 34. Fleet ME, Liu X, King PL. Accommodation of the carbonate ion in apatite: an FTIR and X-ray structure study of crystals synthesized at 2–4 GPa. Am Mineral 2004;89:1422-1432.Article
- 35. Pezzatini S, Solito R, Morbidelli L, Lamponi S, Boanini E, Bigi A, et al. The effect of hydroxyapatite nanocrystals on microvascular endothelial cell viability and functions. J Biomed Mater Res A 2006;76:656-663.ArticlePubMed
- 36. Dos Anjos S, Mavropoulos E, Alves GG, Costa AM, de Alencar Hausen M, Spiegel CN, et al. Impact of crystallinity and crystal size of nanostructured carbonated hydroxyapatite on pre-osteoblast in vitro biocompatibility. J Biomed Mater Res A 2019;107:1965-1976.PubMed
- 37. Ma G.. Three common preparation methods of hydroxyapatite. IOP Conf Ser Mater Sci Eng 2019;688:033057.ArticlePDF
- 38. Ghasemi M, Turnbull T, Sebastian S, Kempson I. The MTT assay: utility, limitations, pitfalls, and interpretation in bulk and single-cell analysis. Int J Mol Sci 2021;22:12827.ArticlePubMedPMC
REFERENCES
Tables & Figures
REFERENCES
Citations
Citations to this article as recorded by 
- Smart Nanomaterials: Current State and Future Prospects in Drug Delivery and Tissue Engineering
E. Elizabeth Rani, D. Sakthi Sanjana, E. Karthikeyan, J. Nandhini
Biomedical Materials & Devices.2026; 4(2): 1455. CrossRef - Thermoresponsive Nanomaterials: Revolutionizing Cancer Theranostics
Bellarmin Michael, Mohanakrishnan Srinivasan, Karthikeyan Elumalai, Lokeshwar Ravikumar, Sivaprakash Kathiresan, Nandhini Jayaprakash
Biomedical Materials & Devices.2026; 4(3): 2697. CrossRef - Physicochemical and antibacterial evaluation of novel nano α-TCP–AgNPs biocomposites for direct pulp-capping applications
Selviana Wulansari, Hendra Dian Adhita Dharsono, Nasrul Wathoni, Rosalina Tjandrawinata, Arief Cahyanto, Moehamad Orliando Roeslan
Frontiers in Oral Health.2026;[Epub] CrossRef - Physicochemical effects of nano type-B bone substitute on pulp protective cement formulations
Njwan Fadhel SHEHAB
Dental Materials Journal.2026; 45(1): 92. CrossRef - Recycling waste for sustainability: The green synthesis of silver nanoparticles from Bougainvillea glabra green waste, and the evaluation of their antioxidant, cytotoxic, catalytic, antibacterial and in-silico molecular docking properties
Hafsa Naleem, Mathivathani Kandiah, Beneli Gunaratne, Ominda Perera
Next Research.2026; 11: 101990. CrossRef - Comparative evaluation of compressive strength and morphological interface of carbonated hydroxyapatite with other pulp capping materials: An in vitro analysis
S. Swathi Priyadharshini, Chinnasamy Ragavendran, I. Anand Sherwood, Ramanaramya Jeyapalan
Endodontology.2025; 37(1): 90. CrossRef - Bioactive Dioxo-Phosphobetaines derived from the reaction of Dichlorodinitrobenzofuroxane with various phosphines
Irina V. Galkina, Haiyan Fan, Semen R. Romanov, Dmitriy I. Bakhtiyarov, Luisa M. Usupova, Svetlana N. Egorova, Yulia V. Bakhtiyarova, Enrico Benassi
Bioorganic Chemistry.2025; 163: 108695. CrossRef - Near-infrared laser-activated PLGA-PDA core-shell nanohybrids for synergistic photothermal antibacterial therapy and sustained ion release in orthodontic white spot lesions prevention
Zezhou Feng, Yujiang Liu, Silu Sun, Minmin Si, Di Huang, Zhiyuan Feng
Journal of Dentistry.2025; 162: 106078. CrossRef - Formation and utilization of soluble microbial products in denitrifying biofilters at different carbon-to-nitrogen ratios: Microbial community characteristics
Fangyuan Jiang, Xianyang Shi
Journal of Environmental Chemical Engineering.2025; 13(6): 119554. CrossRef - Bioactivity and biocompatibility of bioceramic-based pulp capping materials in laboratory and animal models
Rafiqul Islam, Md. Refat Readul Islam, Kenta Tsuchiya, Yu Toida, Hidehiko Sano, Monica Yamauti, Hany Mohamed Aly Ahmed, Atsushi Tomokiyo
Journal of Materials Science: Materials in Medicine.2025;[Epub] CrossRef - Physical, Chemical, and Biological Properties of Graphene Nanoparticle-added Tricalcium Silicate Formulations: A Systematic Review
Soundaria Srinivasan, Deepa Gurunathan, Lakshmi Thangavelu
Journal of International Oral Health.2025; 17(6): 453. CrossRef - Advanced structural and compositional profiling of mineral trioxide aggregate incorporated with nano-carbonated hydroxyapatite: a comprehensive X-ray diffraction and energy dispersive X-ray investigation
Njwan Fadhel Shehab, Nadia Hameed Hasan, Alaa Edrees Dawood, Nawal Atiya Khalaf
Biomaterial Investigations in Dentistry.2025; 12: 216. CrossRef
 ePub Link
ePub Link Cite
CiteEvaluation of mineral induction ability and cytotoxicity of carbonated hydroxyapatite for pulp tissue regeneration: an in vitro study
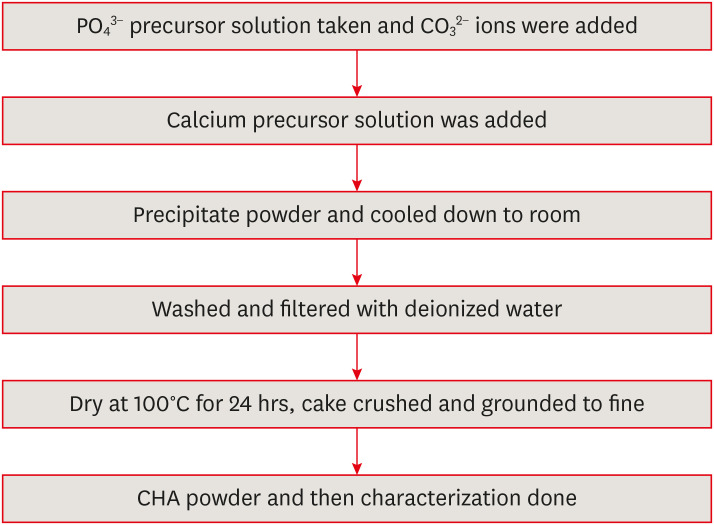
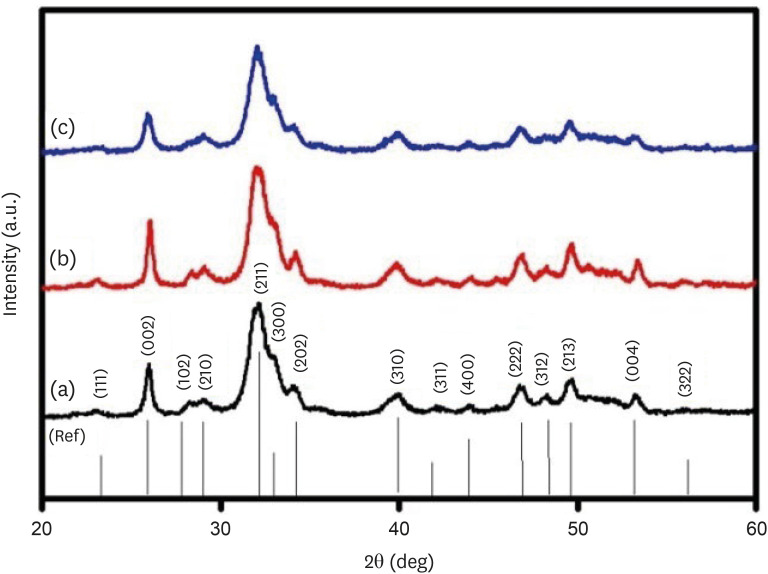
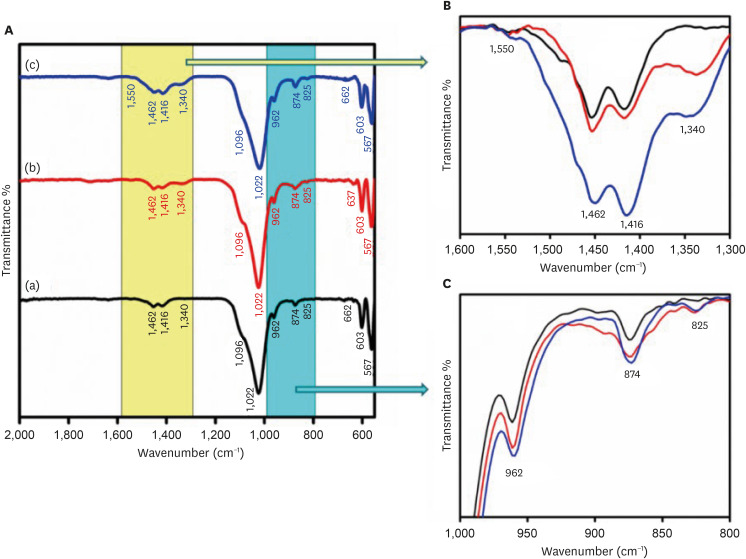
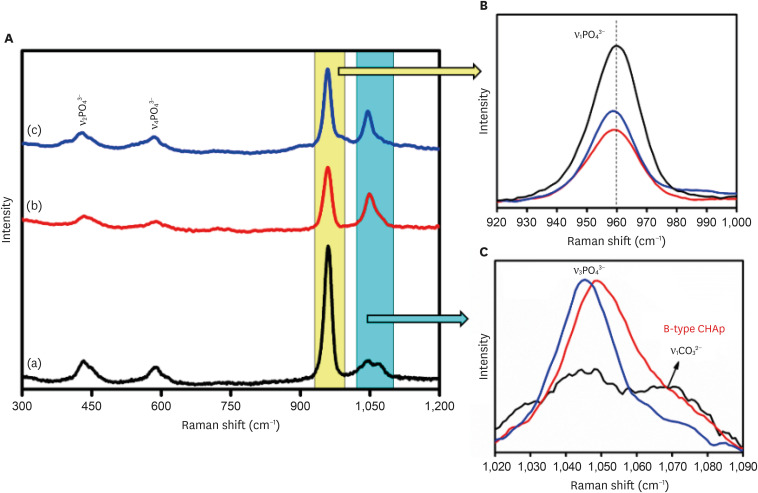
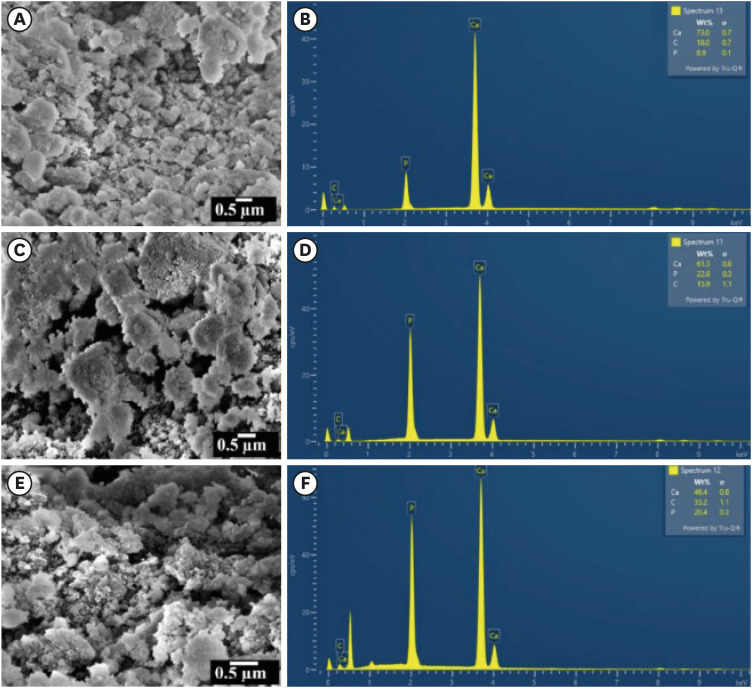
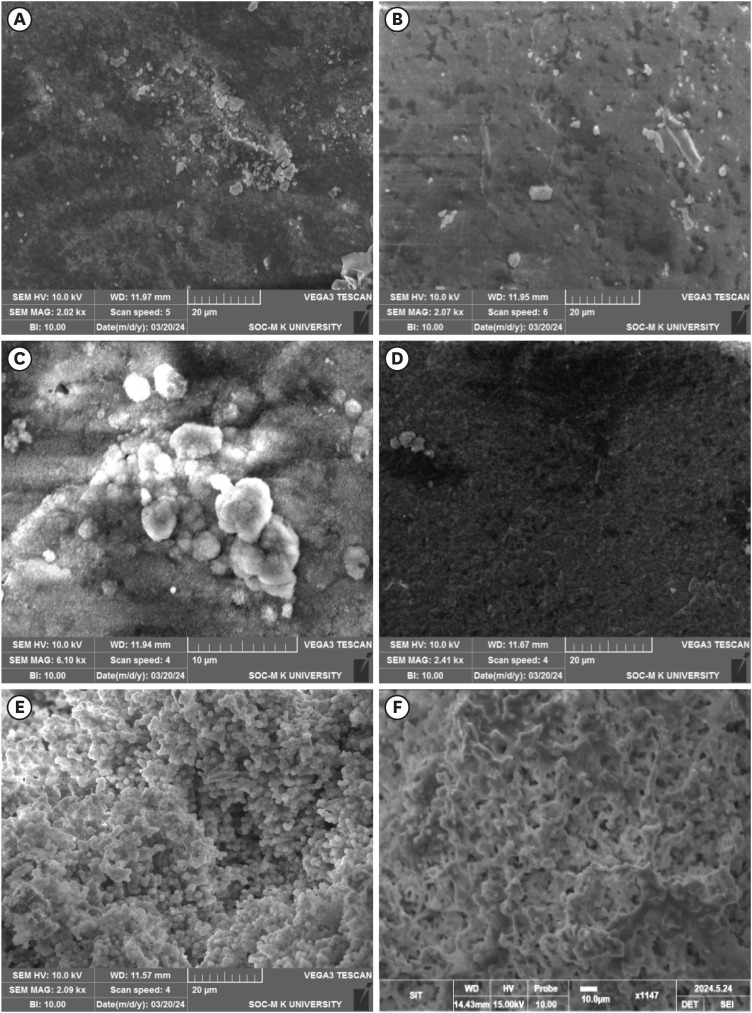
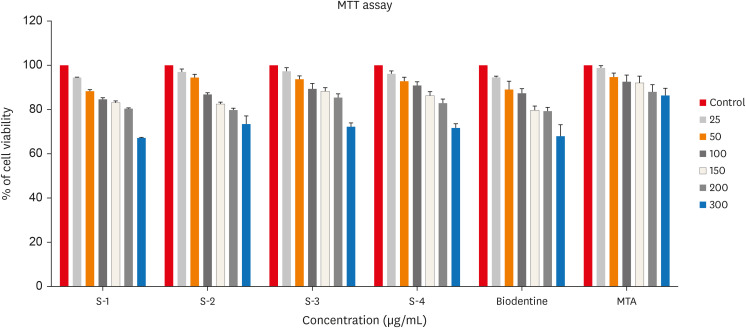
Figure 1 Synthesis of carbonated hydroxyapatite (CHA).
Figure 2 X-ray diffraction patterns of (a) 0.05 M carbonated hydroxyapatite (CHA), (b) 0.1 M CHA, and (c) 0.5 M CHA.
Figure 3 (A) Fourier-transform infrared spectroscopy spectra of (a) 0.05 M carbonated hydroxyapatite (CHA), (b) 0.1 M CHA, and (c) 0.5 M CHA. (B) Magnification of the region 1,600 cm−1–1,300 cm−1. (C) Magnification of the region 1,000 cm−1–800 cm−1.
Figure 4 (A) Raman spectra of (a) 0.05 M carbonated hydroxyapatite (CHA), (b) 0.1 M CHA, and (c) 0.5 M CHA. (B) Magnification of the region 920 cm−1–1,000 cm−1. (C) Magnification of the region 1,020 cm−1–1,090 cm−1.CHAp, carbonated hydroxyapatite powder.
Figure 5 Scanning electron microscopy micrographs and energy-dispersive X-ray analysis spectra for specimens labelled as (A, B) 0.05 M carbonated hydroxyapatite (CHA), (C, D) 0.1 M CHA, and (E, F) 0.5 M CHA.
Figure 6 Scanning electron microscopy micrographs of the simulated body fluid-soaked sample with apatite deposited on the surface of (A) hydroxyapatite, (B) 0.05 carbonated hydroxyapatite (CHA), (C) 0.1 CHA, (D) 0.5 CHA, (E) mineral trioxide aggregate, and (F) Biodentine.
Figure 7
In vitro cytotoxicity of human Dental pulp stem cell lines after culturing in the synthesized carbonated hydroxyapatite (CHA) samples along with positive and negative control determined over 24 hours by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. 0.05 M CHA (S-1), 0.5 M CHA (S-2), 0.1 M CHA (S-3), hydroxyapatite (S-4), Biodentine, mineral trioxide aggregate (MTA).
Figure 1
Figure 2
Figure 3
Figure 4
Figure 5
Figure 6
Figure 7
Evaluation of mineral induction ability and cytotoxicity of carbonated hydroxyapatite for pulp tissue regeneration: an in vitro study
Lattice parameters and crystallite size of (a) 0.05 CHAp, (b) 0.1 CHAp, and (c) 0.5 CHAp
| S. No. | Samples | Lattice parameters (Å) | Average crystallite size (± 1 nm) | |
|---|---|---|---|---|
| a = b | c | |||
| 1 | 0.05 M CHAp | 9.447 ± 0.005 | 6.805 ± 0.004 | 15 |
| 2 | 0.1 M CHAp | 9.389 ± 0.003 | 6.893 ± 0.002 | 12 |
| 3 | 0.5 M CHAp | 9.350 ± 0.006 | 6.923 ± 0.005 | 10 |
CHAp, carbonated hydroxyapatite powder.
Table 1 Lattice parameters and crystallite size of (a) 0.05 CHAp, (b) 0.1 CHAp, and (c) 0.5 CHAp
CHAp, carbonated hydroxyapatite powder.